House dust mites surround us. Burrowing cheerfully into our pillowcases, rugs and furniture, the mites feast on our dead skin cells, breaking them down into small particles they can digest.
Now that your skin is crawling, relax. If you’re like most people, you will never know they are there.
An unlucky minority, however, is very aware of dust mites. Some of these unfortunate folks have a simple dust allergy. But others have an additional condition called atopic dermatitis, often referred to as eczema. They react to the presence of dust — or rather, dust mites — with hideous itching and redness. It wasn’t totally clear what, exactly, caused people with dermatitis to react so badly to dust mites.
It turns out that these people react not to the dust mite, but to its dinner — to the breakdown products of the person’s own skin. The finding helps explain why people with atopic dermatitis react so badly to dust mites, and it provides several new options to help treat the itch. It also resolves a decade-long debate in dermatology — why people with dermatitis are scratching in the first place.
Inside out vs. outside in Atopic dermatitis is known for producing red, cracked and dry skin and, of course, the itching. People usually get diagnosed in childhood. Sometimes it goes away as kids get older, but it still affects between 9 and 30 percent of adults in the United States. Patients with dermatitis who react to dust are told to avoid dusty places and use special pillowcases. For the worst outbreaks, they are often prescribed a steroid cream. In some cases, they can end up in the hospital.
But what causes the itch in the first place? For the past 10 years, scientists have been scratching away at two hypotheses — one called “inside out,” and the other called “outside in.”
The “inside out” hypothesis came first, explains Graham Ogg, a dermatologist with the Medical Research Council Human Immunology Unit at the University of Oxford. The idea was that the immune system was overreacting to normal things: Dermatitis was an inside problem with the immune system itself.
In 2006, however, researchers reported in Nature Genetics that deficiencies in a protein called filaggrin were associated with atopic dermatitis. Now, it’s estimated that 20 to 30 percent of people with atopic dermatitis are also deficient in filaggrin, a protein in the outermost layer of the skin.
“It’s important for moisturizing the skin, keeping the skin hydrated,” explains Ogg. If people with dermatitis are deficient in filaggrin, then “the primary problem isn’t the immune system, it’s the barrier function in the skin.” If the barrier breaks down, more irritants can get in, prompting the immune response and the intolerable itch. So, the “outside in” hypothesis was born. In this view, the immune system wasn’t overreacting; instead it was reacting properly to the avalanche of aggravations it was faced with.
But what if these two hypotheses weren’t at odds, Ogg wondered, and instead were two sides of the same coin? To find out, Ogg and his group began by looking at a molecule called CD1a. This molecule is produced in the skin, and specializes in presenting bits of foreign matter to T cells — the immune system responders that mount attacks against foreign invaders.
It turns out that the CD1a molecules responded to extract-of-house-dust-mite — the delightful concoction that people get scratched with when they are tested for a dust allergy. And when they react, it’s because of CD1a molecules.
To find out if people with dermatitis had more CD1a than people without the condition, the scientists used suction to give eczema sufferers and healthy volunteers large blisters on their arms. The blisters were harvested for their skin and blood cells. And in patients with atopic dermatitis, those skin and blood cells were stuffed with CD1a, far more than in healthy controls.
But what was the CD1a reacting to? Usually CD1a senses fat molecules, presenting bits of them to the immune system to prep it for attack. Ogg and his group assumed that if they analyzed house dust mites, they would find the lipid or fat responsible. Not quite. Instead, they found a protein called phospholipase A2. Phospholipase is an enzyme that dust mites produce that breaks down skin cells, producing fat molecules the mites can digest. CD1a, it turns out, responds to those lipids — reacting to the house dust mite’s dinner. Reacting, really, to the breakdown products of human skin.
This seems like support for the “inside out” hypothesis. CD1a is part of the immune system, and the immune system does seem to be over-reacting.
Filaggrin also had a role to play. The protein doesn’t just create a barrier to keep the skin moisturized — it’s also anti-inflammatory, Ogg’s group showed. If a skin sample was challenged with essence of dust mite, adding filaggrin could damp down the immune response. But eczema patients with low or no filaggrin had no defense. Their skin was more permeable, and there was nothing to stop the inflammation. The “outside in” hypothesis —the idea that the barrier function is the broken part of the system – is true too. Ogg and his colleagues report their findings February 10 in Science Translational Medicine.
“It links together the observations very nicely,” says Muzalifah Haniffa, a dermatologist at Newcastle University in England. It never was a matter of “inside out” or “outside in.” The two are inextricably linked.
Eat like a dust mite, sting like a bee? So, to recap: As dust mites chow down on human skin, they cause damage to the cells. People with dermatitis have immune systems that detect the products of the damage and react, causing itching and pain. Filaggrin, when present, can tamp down the response. But when absent, nothing stops the itch.
The study shows filaggrin is far more than a simple barrier protein. Instead it directly affects immune responses in the skin, something that’s never been seen before, Haniffa notes.
This isn’t the first time that Ogg’s group has come across phospholipase A2. “Bee venom also contains phospholipase. In fact it contains massive amounts,” Ogg explains. Knowing that bee venom and dust mites have something in common helps scientists to understand one of the ways that the immune system senses damage to skin — and gives them another option to consider for treatment.
Right now, clinical trials are focused on stopping the inflammatory proteins produced further down the line. But, Haniffa says, scientists might try methods to increase the amount of filaggrin in the skin — beefing up the barrier against dust mite incursions and reducing the immune response at the same time. Other drugs or creams could target phospholipase A2, inactivating it. Without phospholipase, dust mites wouldn’t be able to break down skin cells, halting any immune reaction.
And that means we can hope for a new day. One with, hopefully, no itch.
A conspicuous “chirp” heralded the first detection of gravitational waves. But some future measurements could be more like hushed murmurs.
Scientists may soon be able to tease out a faint signal of gravitational waves from black hole collisions too distant to be detected directly, scientists with LIGO, the Advanced Laser Interferometer Gravitational-Wave Observatory, report in the April 1 Physical Review Letters. A detection could come in as few as three years — considerably faster than scientists had dreamed possible, the new analysis suggests. When LIGO detected the stretching and squeezing of spacegenerated by a pair of merging black holes, scientists were wowed (SN: 03/05/16, p. 6). The signal stood out well above spurious bumps and wiggles in the data, which are ever-present in LIGO’s extremely sensitive detectors. It rose swiftly in frequency — when converted to sound waves, it was reminiscent of a bird’s chirp — a hallmark of the black holes’ inward-spiraling cosmic dance.
But such obvious swells are outnumbered by a sea of smaller ripples. With these ripples, “you’re looking at black holes which are much farther away,” says LIGO spokesperson Gabriela González of Louisiana State University in Baton Rouge.
LIGO is not sensitive enough to detect these waves outright, but by comparing the data recorded by LIGO’s separate detectors — one in Louisiana and one in Washington state — scientists could identify patterns revealing the presence of the background waves. Such a measurement would allow scientists to compare black hole populations of different ages and could help nail down the conditions under which black hole pairs form.
“My honest opinion was, ‘I’m going to be lucky if we see this result in my lifetime,’” says physicist Emanuele Berti of the University of Mississippi in Oxford, who is not involved with LIGO. He has changed his tune. “Nature was good to us, and now we think that we’re going to be able to see them pretty soon.”
That’s because new estimates of the rate of such black hole mergers are higher than many scientists expected. Using models of binary black hole populations combined with LIGO data, scientists find that LIGO could be sensitive to nearly 2,000 such black hole mergers a year, meeting their most optimistic predictions.
The possibility shows LIGO’s versatility, González says. “There are good prospects of all kinds — it’s not just detections of single events.”
For centuries, the mouth and the body have been disconnected — at least when it comes to health care. Through the Middle Ages and beyond, teeth fell under the care of barbers, who could shave a customer and pull a molar with equal skill. In the 1700s, French surgeon Pierre Fauchard published the Treatise on Teeth, establishing dentistry as its own science.
Across the channel in England, as physicians gained stature in the 19th century, surgeons and dentists engaged in a power struggle. In the modern United States, after medicine became linked to employer insurance and Medicare, the fissure between medicine and dentistry widened. Insurance coverage began at the throat. So when Salomon Amar, a periodontal specialist at Boston University, began exploring links between oral bacteria and heart disease in animal studies in the late 1990s, reactions were lukewarm. “Many cardiologists thought we were a bit crazy,” he says. Skepticism still abounds, but the same molecular tools that have dramatically changed understanding of the gut microbiome are now allowing scientists to track and examine bacteria in the mouth. Advocates of a connection between the artery disease atherosclerosis and microbes are hoping to find convincing proof of their suspicions, while exploring links between ailing gums and other conditions, including cancer, arthritis, diabetes and even Alzheimer’s disease.
The work has profound implications for public health, given that more than 65 million American adults are thought to have periodontal disease, which occurs when bacterial overgrowth inflames the gums and can lead to erosion of gums and bone. If it turns out that periodontal decay drives other diseases, doctors would have a new, and relatively simple, means of prevention.
Wenche Borgnakke, a dental researcher at the University of Michigan in Ann Arbor, has been making this case for years, citing “solid evidence that periodontal treatment has an effect on systemic disease.” She points to a study published last year in the journal Medicine comparing patients on dialysis who received periodontal treatment with those who did not. Those getting treatment had an almost 30 percent lower risk of pneumonia and hospitalization from infections. Another study published earlier this year found that gum disease is associated with a roughly 10 percent higher mortality over 10 years among patients with kidney problems. Researchers working in the field often point out that about half of all deaths from atherosclerosis occur in people who do not have any classic risk factors, such as high cholesterol or obesity. Something else — something as yet unknown — is also contributing to heart disease. Even the root cause of many cancers is largely unexplained. Most women with breast cancer, for instance, have no risk factors other than older age. Says Jean Wactawski-Wende, a cancer epidemiologist at the State University of New York at Buffalo: “The more I work on oral health and cancer, the more I think, ‘Oh my gosh, I’ve got to keep my teeth clean.’ ”
Foul mouth To date, more than 500 scientific papers have weighed in on the connection between atherosclerosis and gum disease. Officially, the theory remains “biologically plausible,” but unproven, according to the American Heart Association’s formal position. Some concepts are undisputed: For one, the microbes that live in the mouth don’t stay in the mouth. The simple act of brushing allows bacteria clinging to the teeth and gums to leak into the bloodstream.
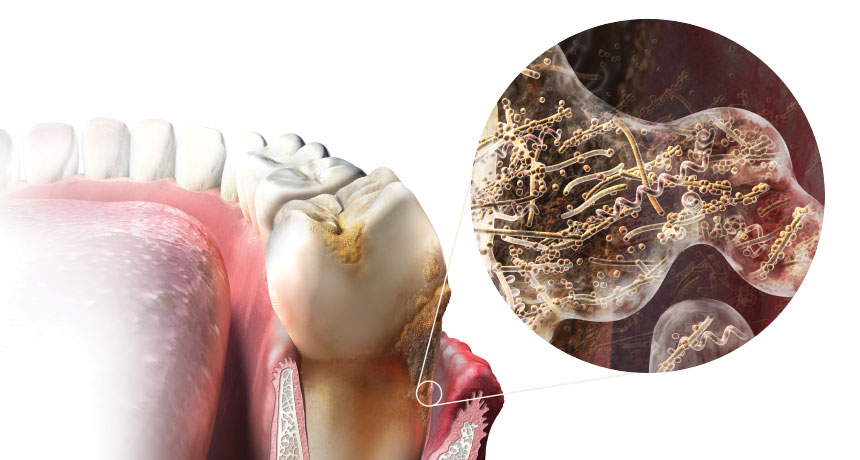
As the posters at the dentist’s office attest, neglected oral hygiene encourages bacterial growth, allowing the microbes to breed on and between teeth, as well as under the gums. What the illustrations don’t show is that these microorganisms form a biofilm, a tough microbial community bound together with sugar molecules in a thin coating. This is the plaque your dentist warns you about.
“If you do not brush your teeth, it will sit there and accumulate. As that plaque gets thicker and thicker, there is less and less oxygen in the deepest layers,” Borgnakke says. Safely sheltered, the innermost plaque starts to favor anaerobic bacteria, which, when they escape into the blood, can survive in the oxygen-starved nooks and crannies deep inside the body.
As plaque builds up, gums get irritated, swell and draw more blood into the distressed tissue. Eventually, chemicals produced by the biofilm break down the thin layer of cells that form a boundary between the gums and the blood vessels. Periodontitis officially occurs when gum and bone tissue starts to deteriorate. The space between the tooth and gums forms a pocket; dentists measure the depth of the pockets to determine the severity of infection. “We usually think of an infection as some bug from the outside that attacks the body,” says Borgnakke. “In this case, it’s an internal infection.”
It was once thought that only a handful of microbial species were involved in the development of periodontitis, but the latest studies have revealed that many of the microbes responsible for gum disease come from “previously underappreciated species,” according to a 2015 report in Advances in Experimental Medicine and Biology. Because many bacteria resist growth in a laboratory, only a small portion of some 500 to 700 species of oral microbes have been well characterized.
One aggressive pathogen, an organism called Porphyromonas gingivalis, has antennae that stick out and can pry open the space between two cells, Borgnakke says. “This is a really, really nasty bug.” Within minutes of invading blood vessels, P. gingivalis and its gang of accomplices are ferried to distant sites, where they can set up outposts. “Bacteria that normally live in the mouth are found in every organ in the body, and even muscle cells,” she says.
The body doesn’t take this assault lying down. The immune system gets agitated and tends to stay in a state of slow simmer. But the bacteria that cause periodontal disease have a knack for turning the body’s defense on its head, according to a 2015 review in Nature Reviews Immunology. Case in point: Common white blood cells called neutrophils are deployed to the failing gums — where they not only fail to control the infection, but also end up releasing enzymes that further destroy tissue. The immune system also releases an avalanche of chemicals designed to help control the infection. For example, the liver starts producing C-reactive protein, a molecule that has such an important role in signaling the rise of heart disease that it is considered a risk factor by some researchers.
Smoking gums Even after two decades of study, it has been hard to directly link periodontal dynamics to blocked arteries, despite hundreds of studies that have tried. There are seemingly smoking guns. Among them, P. gingivalis is commonly found lodged inside arteries, and the development of plaque in the arteries is driven by many of the same inflammatory chemicals triggered by periodontal disease. Many researchers also point to C-reactive protein, which is probably present long before atherosclerosis develops. But people with periodontitis also tend to share well-known risk factors for heart disease, such as high cholesterol, smoking and obesity. A sugar-sweetened diet that promotes oral decay is no friend to your arteries. The relationship is also hard to study because both atherosclerosis and periodontitis unfold slowly over time, so epidemiologists must rely on indirect measures of disease.
Experts line up on both sides. “If there is an association, it’s a very weak one,” says Peter Lockhart, former chairman of oral medicine at Carolinas HealthCare System in Charlotte, N.C. An expert on heart valve infections, Lockhart was one of the leaders of an American Heart Association panel that reviewed the evidence before releasing an official statement in 2012. “I think the question has been answered for now,” he says. For cardiologists, the threat from periodontal disease “pales by comparison to the known risk factors that need to be focused on.”
Others aren’t ready to abandon the hypothesis. In 2015 in the journal Atherosclerosis, a team of German researchers reviewed studies released since the AHA statement. They pointed out that a large body of work published in the previous three years, using more refined tools and study design, shows that a connection between the two “cannot be ruled out.” One study, published in PLOS ONE in 2014 from researchers at the University of Florida in Gainesville, Meharry Medical College in Nashville and elsewhere, claims to have found a causal relationship, at least in mice. A significant portion of animals that drank water containing P. gingivalis experienced inflammation and bacterial accumulation in their hearts and blood vessels. Very few unexposed animals did.
Into the brain While the artery studies carry on, new research is finding oral bacteria in surprising places. The brain, for one. In 2013, a team of researchers from Florida and the United Kingdom compared brain tissue samples from 10 people who had died from Alzheimer’s disease with samples from 10 people who had died from other causes. Signs of P. gingivalis infection showed up in four Alzheimer’s patients but in none of the comparison patients, the researchers reported in the Journal of Alzheimer’s Disease. In a follow-up experiment published in the same journal, the researchers inoculated P. gingivalis into the mouths of 12 mice genetically protected from Alzheimer’s. Six months later, evidence of the same bacteria appeared in the brains of three-fourths of the animals. Another type of oral bacteria, spirochetes called Treponema denticola , “are already known to enter the brain,” says neuroscientist Sim Singhrao of the University of Central Lancashire in England. Traveling along the nerves that connect to the jaw, “they are a bit like jellyfish, crawling up into neurological tissue.” Once nestled inside the brain, oral bacteria could trigger an inflammatory chain reaction that eventually destroys certain nerve cells and leads to Alzheimer’s disease, says StJohn Crean , Lancashire’s executive dean of the College of Clinical and Biomedical Sciences. He points out that Chinese researchers, writing last year in the Journal of Periodontal Research, found that people carrying certain versions of APOE, a gene linked to Alzheimer’s, were also more likely to suffer aggressive periodontal infection. Finally, a study published in March in PLOS ONE found that among 59 people with hallmarks of Alzheimer’s disease followed for six months, those with periodontitis experienced cognitive decline at more than six times the rate as those without gum disease.
“We’ve moved on from that ‘this-can’t-be-right’ feeling,” Crean says. He is hoping to get funding for a study that would compare progression of Alzheimer’s among people who receive intensive oral hygiene, such as frequent dental-office–style cleanings, compared with those who brush and floss regularly. But he also notes that the arrow connecting gum disease and Alzheimer’s could point in both directions. “When your memory goes, you’re not going to remember to brush your teeth.”
Teeth and tumors Providing still more reason to invest in dental floss, new research is raising questions about cancer’s link to gum health. Aside from oral cancers, the cancer connection was barely on the scientific radar before 2008, when a study appeared in Lancet Oncology. Some research had suggested that gum disease is associated with higher cancer mortality, but questions remained about the influence of smoking. In the study in Lancet Oncology, researchers from Imperial College London, Harvard Medical School and elsewhere reviewed data for almost 50,000 men enrolled in the Harvard Health Professionals Follow-Up Study. That study found a small increased risk of cancer mortality in men with periodontal disease.
A second study, published in February in Annals of Oncology, found that men with advanced periodontal disease who had never smoked nonetheless had a 2.5 times higher risk of cancers associated with smoking, such as lung, bladder and esophageal tumors. The researchers hypothesize that gum disease might trigger the same sort of immune response that tobacco does. Another study examined data from more than 73,000 participants of the Women’s Health Initiative, which gathered health information from volunteers over 15 years. Participants diagnosed with periodontal disease had a 14 percent increased risk of breast cancer compared with women with healthy gums. “It’s a modest increase, but when 50 percent of adults are diagnosed with periodontal disease, you could see this becoming a very important factor for prevention,” says Buffalo’s Wactawski-Wende, who led the study, published in January’s Cancer Epidemiology, Biomarkers & Prevention.
Laboratory studies are also offering compelling evidence of associations with certain cancers. Almost a dozen studies conducted over the last five years have found one particular species of mouth bacteria, Fusobacterium nucleatum, living in seeming abundance in colorectal tumors. Like P. gingivalis, F. nucleatum thrives in diseased gums and in low-oxygen areas. Wactawski-Wende is studying samples of various tumors to look for oral organisms. Burning questions
Given that periodontal disease causes the immune system to remain in a state of irritation, other lines of research have tried to tie diseased gums to inflammatory diseases like rheumatoid arthritis and diabetes. Writing last year in the journal Mediators of Inflammation, researchers from the University of Ceará in Brazil reviewed published studies on rheumatoid arthritis, concluding that “the majority of the articles have confirmed that there is a correlation,” especially among women. Both gum disease and arthritis, they wrote, could even feed off one another, amplifying a hyperactive immune system that makes both conditions worse.
A long line of research has also examined the relationship between diabetes and periodontal disease. In 2013, Borgnakke and an international team reviewed the evidence in the Journal of Clinical Periodontology. Of the 17 studies they found to have sufficient quality, the evidence suggests that people with poor periodontal health have a greater chance of developing early symptoms of diabetes and having greater complications from the disease once it develops. But she acknowledges that diabetes, and in fact all conditions under study, have multiple causes, making the role of any one culprit difficult to determine.
It’s also hard to account for the role of genetics. “You could have two patients with the same amount of plaque. One patient will have really deep pockets [between teeth and gums], and the other one will have no consequences,” she says. “That’s why it’s so hard to say anything in general.”
Even as research continues, those involved concede that they may never satisfy skeptics, given the slim chance of ever having a long-term prospective study. That research would need to monitor the cardiac health of a large population over an extended time, half with gum disease and half without, to determine if those with periodontal problems experienced worse cardiac health. But given the length of time it takes for both gum disease and systemic disease to reveal themselves, a study would need to involve thousands of participants over many years to be definitive, Amar says. “It would be financially prohibitive.” And he points out that pharmaceutical companies, which often help fund large clinical trials, would not back a study that has no product for them to eventually sell.
“Causality may not ever be demonstrated,” he says. To most doctors, the mouth will probably remain unconnected to the body. Amar and others will nonetheless continue, in hopes their work may one day give health professionals a little more to chew on.
Imagine that you’re walking through a secluded forest somewhere in Southeast Asia. You’re far from modern conveniences, like electricity. The trees overhead block out much of the light. It’s quiet, except for the sounds of ruffling leaves or the calls of birds. Suddenly, you’re struck by the scent of popcorn.
You’d probably think you were going nuts. But you might have just picked up the trail of a bearcat, also called a binturong, a rare carnivore that sports a bushy, prehensile tail and is most closely related to the civet. And the animals’ popcorn scent is so pronounced that many zookeepers name their bearcats after the buttery movie snack.
The source of the scent, though, has been a mystery. Now Lydia Greene of Duke University and colleagues have found that the chemical that gives popcorn its delightful scent, 2-acetyl-1-pyrroline (2-AP), is also found in bearcat pee. They report their finding in the June issue of The Science of Nature.
As anyone who has ever owned a dog will know, urine is a key way that carnivores mark their territory. But the chemicals inside urine can also help an animal advertise who they are and if they’re ready to mate, as well as provide ways for a group of animals to maintain hierarchies or bonds between individuals.
Bearcats, like other carnivores, leave these chemicals wherever they go — but they’re a bit more aggressive about it than your pet poodle. A binturong pees in a squat, which soaks its feet and tail in smelly urine that is left behind with every step. Greene’s team investigated the composition of the animal’s pee to see how chemicals might influence how females maintain their social dominance over males. Of the 29 chemicals they detected in the urine of 13 female and 20 male bearcats living in captivity at the Carolina Tiger Rescue in Pittsboro, N.C., only one could be found in the pee of every animal: 2-AP.
The chemical, the researchers write, “likely explains the binturong’s signature scent,” and “may serve as a species identifier.”
Just where the chemical is coming from, though, remains unknown. The researchers checked the animals’ food and didn’t find any traces of 2-AP. One possibility is that bacteria in the bearcats’ gut or elsewhere are responsible. Bacillus cereus, commonly found in the intestines of vertebrates, is known to synthesize 2-AP.
But the chemical probably isn’t getting into bearcats in the same way as popcorn. That’s because 2-AP isn’t found in popcorn until the kernels have undergone the high heat necessary to pop the corn — an unlikely activity for jungle-dwelling mammals.
Rachel Dutton’s research is cheesy, by design. The microbiologist at the University of California, San Diego uses cheese rinds to study how microbes form communities.
Dutton, who has a long-standing interest in how bacteria and other microbes interact, got the inspiration for her studies several years ago while visiting the Marine Biological Laboratory in Woods Hole, Mass. In the salt marshes there, multiple species of bacteria, archaea and other microbes were growing in thick, many-layered mats. They would have been perfect for studying microbes in groups. Except for one thing: Many organisms that thrive in those mats won’t grow in captivity. Dutton needed a microbe community that she could pick apart, manipulate and reconstruct in the lab. The solution came with a round of cheese from France. Sliced open, its rind reminded Dutton of the microbial mats on the coastal salt marshes. “I took a piece of cheese into the lab and put it under the microscope,” she says. “Everything I was looking for in a microbial community was present.” Dutton and colleagues did genetic analyses of 137 cheeses from 10 countries and identified 24 genera of bacteria and fungi that are common in cheeses and will grow in the lab, the team reported in 2014 in the journal Cell.
By sampling a Vermont cheese as it aged over 63 days, the group also discovered that rind communities don’t form instantly. At the beginning, community members included Proteobacteria and Leuconostoc bacteria, plus candida yeast commonly found in raw milk. Within a week, Staphylococcus had overwhelmed the Proteobacteria. As the cheese ripened, Brevibacterium and Brachybacterium plus Penicillium and Scopulariopsis fungi became prominent inhabitants of the rind. That pattern held whenever those organisms congregated in a cheese in the lab.
Cheesemakers from Vermont taught Dutton how to ferment cheese curds and create her own lab version of a dry-aged cheddar. Although cheeses can have complex combinations of microbes — stinky cheeses have the most diverse mixes — Dutton’s lab crafts a more simplified rind using three types of fungi and four bacteria. The researchers grow the microbes in pairwise combinations to learn how they interact. Studying cheeses and other fermented foods could teach scientists how microbial communities evolved in different places and lead to the creation of new, tastier and safer foods, Dutton and Benjamin Wolfe of Tufts University wrote last year in Cell.
There’s one drawback to the cheesy research: The lab has a ripe odor, Dutton says. “We look like a normal microbiology lab, but we don’t smell like one.”
Hair standing on end during a thunderstorm is a bad sign — it means lightning is on the way. On exoplanet HAT-P-11b, though, static hair might be the least of your worries. A barrage of lightning striking 530 times as often per square kilometer as storms in the United States could be the cause of a surge of radio waves detected from the planet several years ago, Gabriella Hodosán of the University of St. Andrews in Scotland and colleagues suggest online April 23 in Monthly Notices of the Royal Astronomical Society.
In 2009, astronomers recorded radio waves coming from the HAT-P-11 system that ceased when the planet slipped behind its star, suggesting the planet was the source of the signal. A second look in 2010 found no radio waves.
Scientists have detected lightning on planets closer to home, including on Venus and Jupiter, but not on a planet orbiting another star. HAT-P-11b is too close to its star for astronomers to see visible flashes of light. But an infrared telescope might pick up a stockpile of hydrogen cyanide created by the electrical discharge.
Solar outbursts may have supplied early Earth with the right stuff for life.
Based on telescope observations of young sunlike stars, researchers estimate that “super” solar flares bombarded Earth with energetic particles at least once a day around 4 billion years ago. Collisions between the particles and molecules in Earth’s atmosphere produced nitrous oxide, a planet-warming greenhouse gas, and hydrogen cyanide, a crucial component for building DNA, the researchers propose May 23 in Nature Geoscience.
Those chemical products warmed and fostered emerging life, says study coauthor Vladimir Airapetian, an astrophysicist at NASA’s Goddard Space Flight Center in Greenbelt, Md. “Our sun, worshipped by ancient civilizations, wasn’t just a source of warmth, it also produced ingredients for life,” he says. Life’s earliest traces date back to around 4.1 billion years ago (SN Online: 10/19/15). At the time, the sun was about 25 to 30 percent dimmer than it is today — too faint to keep Earth’s temperatures above freezing without the help of additional greenhouse gases (SN: 5/4/13, p. 30). Simulating the early sun using star data collected by NASA’s Kepler space telescope as a guide, Airapetian and colleagues found that while dim, the sun was probably wilder in its youth. Solar flares, also called coronal mass ejections, probably erupted more often and with more ferocity, producing storms 1,000 times as powerful as the most intense flares on record.
Those storms made a big impact on Earth, the researchers propose. The storms temporarily squeezed the magnetosphere, the protective magnetic bubble surrounding Earth, to one-sixth its normal height. That squashing allowed more solar particles to rain into the atmosphere. The dive-bombing particles ionized and broke apart nitrogen molecules in the air. Those molecules reassembled into new ones such as hydrogen cyanide, which can produce DNA bases and amino acids.
Another product, nitrous oxide, is a greenhouse gas nearly 300 times as potent as carbon dioxide. The additional nitrous oxide could have kept Earth from freezing during the sun’s dim days, the researchers propose.
The solar flares would have impacted more than just Earth. Similar effects would also have occurred on Mars, Airapetian adds, potentially improving the Red Planet’s habitability.
The interactions create the right molecules but in the wrong place, says Ramses Ramirez, an astrobiologist at Cornell University, who wrote an accompanying perspective piece in the same issue of Nature Geoscience. The flare-formed molecules would have originated in the upper atmosphere, not near the surface where most greenhouse warming takes place and where life would have taken root. While the idea is plausible, some other mechanism is needed to “get the molecules down so the critters can utilize them,” he says.
The influenza virus is a quick-change artist. In a few decades, its genome can evolve as much as animal genomes can over millions of years. That means that the viral proteins, including those that alert our bodies to an infection, constantly reinvent themselves, threatening our immune systems and frustrating vaccine developers.
For Jesse Bloom, a biologist studying how evolution affects proteins, that relentless change is an opportunity. Thanks to data collected during past flu seasons, Bloom knows the exact genetic makeup of some ancestors of today’s influenza viruses. His lab group at the Fred Hutchinson Cancer Research Center in Seattle uses that information to figure out how the viruses made their immunity-dodging transformations.
Bloom and others are part of a growing group of scientists who practice “evolutionary biochemistry.” They seek to explain life’s tremendous diversity and determine exactly how that diversity emerged. Rather than focusing on how plants or animals adapted to different environments, however, these researchers consider diversity on a much smaller scale: Their work aims to explain how the small set of proteins that powered primitive life-forms evolved into the millions of specialized proteins that drive biological processes today.
Exploiting the genetic records, Bloom can assemble virus proteins that existed in bygone times, then reconstruct how they evolved, one amino acid at a time. Other researchers are analyzing modern species to resurrect the ancestral forms of biological molecules that have evolved over millions of years.
With a historical protein in hand, researchers can test how swapping out a single amino acid — as evolution might have done — changes how the protein flexes or folds and connects (or doesn’t) with other molecules. By trying out alternate versions of a protein’s history through stepwise amino acid changes, scientists can learn how a protein’s physical form has both enabled and constrained its evolution.
Ultimately, this work might answer some long-standing questions: To what extent does evolution depend on chance events? Can evolution reach the same point by traveling different paths? How does biological complexity evolve? Such experiments are also helping researchers who study modern proteins sort out how the order of amino acids relates to biological function. That ordered series of amino acids is spelled out by the gene that holds the blueprint for a protein. Once the proper amino acids are strung together, they origami-fold into tiny structures with nooks and protrusions that determine what the protein does inside a cell. A protein’s folded shape lets it grab on to specific bits of DNA or hasten certain chemical reactions. Mutations in a gene can shift the resulting protein’s shape or alter subtle aspects of its behavior so that, over time, a protein’s function can change. But the possibilities are not endless. New proteins that fall apart, fail to fold or don’t perform as needed don’t survive the tests of natural selection.
“The physical determinants of folding, stability, solubility, function and specificity are absolutely essential aspects of the evolutionary process,” says University of Chicago biologist Joe Thornton. “That has not been widely appreciated or explicitly addressed until pretty recently.” Now, Thornton says, it’s clear that to understand molecular evolution, it’s important to study proteins as functioning, physical objects.
As they reconstruct proteins’ pasts, researchers are finding that genetic mutations sometimes remodel a molecule just enough to give a chance to other mutations that would have failed earlier. That creates opportunities for new features and functions to evolve — an idea that biologists have considered for decades but have only just begun to explore in the lab.
Bloom and colleagues, for instance, used an influenza virus protein called nucleoprotein to examine how interactions among mutations have affected the overall evolution of the virus. Understanding the combined effects of several mutations could allow researchers to anticipate the short-term effects of new genetic variation. That knowledge could help improve forecasts of which viral strains are likely to circulate in upcoming flu seasons, important information for designing effective vaccines.
Comparing nucleoprotein genes from strains of the virus isolated in 1968 and 2007, Bloom’s team mapped out the most likely steps by which the 1968 protein morphed into its newer form. Though nucleoprotein still plays the same role that it did in 1968 — aiding in the assembly of viral RNA — 33 of its 498 amino acids changed over those four decades, and a few changed more than once, the researchers reported in 2013 in eLife.
Bloom’s team built the 1968 nucleoprotein, then tested the effects of introducing each historical mutation. Some of the mutations affected parts of the protein that tip off a person’s immune cells that an invader is present — they probably helped the flu virus avoid detection. But on their own, some of those changes were bad for the virus: The nucleoprotein could no longer stay properly folded long enough to do its job.
During the course of the nucleoprotein’s evolution, some mutations boosted the protein’s stability, giving it a bit of a buffer. When later mutations occurred, allowing the virus to buck immune recognition, these earlier changes probably held the structure stable so the protein could still function.
When a mutation’s effects depend on other mutations, this interplay is called epistasis. These interactions within individual molecules have been important in shaping evolutionary trajectories, says University of Oregon biophysicist Michael Harms, who is studying how diverse functions evolved in a group of proteins called s100s. He calls epistasis “the common feature in all of evolution.”
Codependent interactions don’t occur just between pairs of mutations. They can be significantly more complex. Analyzing data from other labs, Harms has found epistatic interactions involving up to six different mutations. Such interplay means that in many cases, if genes had transformed themselves just a bit differently, evolution would have veered onto a different course.
Green light Scientists call mutations that lay the groundwork for future change “permissive” mutations. Some protein functions came about only after permissive mutations modified an evolving molecule in highly improbable ways.
Thornton uses ancestral protein reconstruction to study how steroid hormones — which control stress responses, growth and sexual developmental in vertebrates — evolved partnerships with their receptors. Receptors are proteins that bind to specific partners to activate responses in the cell. By comparing steroid receptors in different species, Thornton can map the evolutionary relationships between the molecules and infer the likely amino acid sequence of their common ancestor. Then he introduces a DNA molecule that encodes the long-extinct protein into lab-grown cells. Those cells use the genetic instructions to manufacture a tiny piece of the deep past.
Many of Thornton’s studies begin with a 450-million-year-old receptor protein that he and colleagues reconstructed in 2006. The protein gave rise to modern receptor molecules that are activated by different hormones. One receptor, the glucocorticoid receptor, responds to the stress hormone cortisol. The other, the mineralocorticoid receptor, controls levels of salt and other electrolytes in response to the hormone aldosterone. Thornton’s team found that their reconstructed ancestor could be activated by both cortisol and mineralocorticoids.
A receptor that responded only to cortisol appeared 40 million years after the promiscuous receptor, Thornton showed. His team found a set of amino acid changes that converted the general ancestral receptor into the cortisol-specific one. But the mutations that changed the ancient receptor’s preference couldn’t have generated a functional receptor by themselves, experiments showed.
“The function-switching mutations are not tolerated on their own,” Thornton says. They destabilize parts of the receptor. Like the flu virus’s evolving nucleoprotein, the ancestral receptor’s structure had to be buttressed before it could withstand the mutations that would make the receptor choosier.
Two amino acid changes quietly readied the ancient receptor for its transformation, Thornton and colleagues reported in 2009 in Nature. Without them, the path to the function-switching mutation would have been inaccessible. “If we were to wind back the clock and set history rolling again, it’s very unlikely that those permissive mutations would occur,” he says. “We would have ended up with a very different glucocorticoid receptor and a very different endocrine system.”
Thornton and Harms, then a postdoctoral researcher in Thornton’s lab at University of Oregon in Eugene, explored whether evolution could have taken an alternate route to the same end. Harms created and screened thousands of variants of the ancestral protein, searching for alternative mutations that might have set it up for the same functional switch. He found none, the researchers reported in Nature in 2014. Evolution, it seems, had acted on a rare opportunity.
Biophysical analyses of variant receptor proteins showed why so few mutations enabled cortisol-specific binding to evolve. Although certain parts need extra support, the receptor also needs to be able to transition between two forms: an inactive conformation when no cortisol is present, and a gene-activating conformation when the hormone binds. Some mutations stabilize the active form of the receptor too much, locking it into an “always-on” configuration. Mutations also had to be compatible with the ancestral protein on their own, before the function-switching mutations were introduced.
“A mutation has to fulfill all these requirements, and that is not easy to do,” Thornton says. “That seems to be the explanation for why permissive mutations [for this functional switch] are so rare.”
But not every new function is the result of complicated epistatic interactions. In January in eLife, Thornton and Ken Prehoda of the University of Oregon described an ancient protein that gained a completely new function by way of a single amino acid change.
The team studied the origins of an animal protein that helps cells orient themselves in space before dividing. Doing so is vital for positioning new cells in the right places within a growing body. Single-celled life-forms had to get this right before multicellular organisms could evolve. Thornton, Prehoda and colleagues focused on a segment of the protein called GK PID (for GK protein-interaction domain), which orients cells by acting as a scaffold during division. The billion-year-old ancestor of GK PID did nothing of the sort. It was an enzyme predecessor to the modern guanylate kinase, which catalyzes a chemical reaction that cells use to make some of the building blocks of DNA. Amazingly, Thornton says, one mutation was enough to transform the ancestral protein from an enzyme to a working scaffold. That surprising result is an example of why developing general theories about the physical principles shaping evolution requires a grasp of the evolutionary histories of a broader collection of proteins.
“Every time people take [a protein] apart, they see a new feature,” Harms says. Fortunately, he says, thanks to faster computers, better software and a growing number of genomes to reference, research on ancestral protein reconstruction is on the rise.
Roads taken While chance events can shift the landscape of evolution’s possibilities, evolving proteins also have some freedom to explore. They can take more than one path to some functions.
Douglas Theobald, a biochemist at Brandeis University in Waltham, Mass., has seen this in his own investigations of an enzyme that many cells use to produce energy without oxygen. The enzyme, lactate dehydrogenase, evolved from structurally similar enzymes not just once, but at least four times in different groups of organisms. By reconstructing the evolutionary events that transformed a similar enzyme, malate dehydrogenase, into lactate dehydrogenase, Theobald and colleagues found that two groups of single-celled parasites came by the same enzyme in different ways. The researchers reported the findings in eLife in 2014 and in Protein Science in February.
The work demonstrates that different genetic backgrounds may steer evolution along different paths in different organisms but still lead to similar outcomes, Theobald says. “Even if there is a lot of epistasis, there’s still lots of different ways to the same function.”
Biochemist Susan Marqusee of the University of California, Berkeley has also found that there’s more than one way for a protein to do something new.
Marqusee collaborated with Thornton’s team to compare how two bacteria, Escherichia coli and the heat-loving Thermus thermophilus, evolved enzymes that do the same job at very different temperatures. T. thermophilus thrives in hot springs, at temperatures that would cause most proteins to fall apart. Biochemists are eager to borrow from nature’s strategies to engineer heat-tolerant proteins but have struggled to find general principles that account for this property. By reconstructing the common ancestor of the RNA-snipping enzyme known as H1 from E. coli and T. thermophilus, Marqusee’s team found out how the bacterial protein takes the heat.
That 3-billion-year-old common ancestor was less stable than the enzyme that T. thermophilus uses today, the team reported in 2014 in PLOS Biology. As the heat-tolerant protein evolved, its stability steadily increased — not because of any one innovation, but by virtue of distinct biophysical strategies at different points in time.
“The physical chemistry doesn’t really matter as long as in the end, they add up to the right phenotype,” Marqusee says. Because evolution was able to take advantage of different amino acids to boost stability in a variety of ways, the enzyme’s growing resilience to hot environments didn’t depend on the chance occurrence of a particular set of mutations.
Foggy future Studies of how proteins have evolved in the past are unlikely to spell out how evolution will proceed in the future. “The emerging picture is that the role of chance is so great that long-term predictions of the future evolution of any protein is a very risky enterprise,” Thornton says. But recent research does offer insights into how and why today’s proteins do what they do. One example comes from Thornton’s work on how the DNA-binding sites on steroid receptors have evolved along with their DNA targets. The hormone-activated receptors act as transcription factors, binding to specific sections of DNA to switch on certain genes. In 2014, Thornton’s team reported in Cell that a bulky amino acid in an ancestral protein prevented the protein from binding to the stretch of DNA favored by many of today’s steroid receptors. The ancestral protein awkwardly bumped up against the DNA, unable to make enough contact to really grab on. The receptor gained its new specificity when mutations ended those obstacles and introduced new clashes that blocked its access to the former binding site.
Researchers often can’t tell which differences between two related proteins make them behave differently. But reconstructing evolutionary paths can point them in the right direction.
Using ancestral reconstruction, Theobald and Brandeis colleague Dorothee Kern studied how Abl, a growth-promoting protein linked to chronic myelogenous leukemia, diverged from the related Src protein. The researchers wanted to know why the anticancer drug Gleevec binds to and shuts off Abl without obstructing Src, even though Src has a very similar structure. Theobald, Kern and colleagues identified 15 amino acids in Abl that are crucial for Gleevec binding. The amino acids influence how the protein transitions between two different configurations (that shape-shifting is disrupted in some patients with Gleevec-resistant cancers). The finding, published last year in Science, suggests that researchers may be able to develop better drugs by considering these conformational shifts.
Some proteins, or parts of proteins, might even be inherently more able to evolve than others. Certain parts of the fast-evolving viral protein hemagglutinin are unusually tolerant of change, Bloom and Bargavi Thyagarajan, who was a postdoctoral researcher in Bloom’s lab, reported in 2014 in eLife. Antibodies against hemagglutinin are the immune system’s best defense against influenza, but the protein is adept at escaping detection.
The researchers used a relatively new method called deep mutational scanning to build and test hemagglutinin proteins with nearly every possible amino acid change — about 10,000 in all — in viruses grown in the lab. In a host, changes that disguise hemagglutinin from the immune system would be advantageous. Even though there was no immune system to hide from in the lab, viruses still survived more changes to parts of hemagglutinin that would be recognized by an immune system than they did changes to other parts of the protein. Bloom and his graduate student Michael Doud reported a more detailed view of the protein and the areas that are more and less likely to tolerate mutations online on bioRxiv.org in April. That’s good for the virus, but bad for people. Hemagglutinin seems capable of accumulating change in the very sites that vaccine developers would like to remain the same. But the finding also suggests that flu vaccines designed to target less mutation-tolerant regions of hemagglutinin might be more likely to protect against the flu from season to season. That’s a strategy some labs are already exploring — targeting the less-evolvable stalk of hemagglutinin’s lollipop-shaped structure.
It’s not yet clear why certain parts of the hemagglutinin protein tolerate change so well; Bloom hopes that studying the mutational tolerance of other proteins will help researchers figure that out.
“We’re never going to be able to predict evolution precisely, because it’s a highly stochastic process,” Bloom says. “But I think we can make better forecasts about many of the evolutionary processes that affect us. These are really challenging problems, but I think we are getting to the point where we can use experiments and molecular understanding to help us think about these processes.”
The world’s known helium reserves just ballooned. Applying gas-finding techniques from the oil industry, scientists uncovered a vast reservoir of more than a trillion liters of helium gas beneath Tanzania. That’s enough to satisfy the world’s helium needs for around seven years, the researchers announced June 28 at the Goldschmidt Conference, a geochemistry meeting being held in in Yokohama, Japan. The find may allay fears that a global helium shortage will hit when the U.S. Federal Helium Reserve — currently the world’s largest helium source — runs dry within the next few years.
While previously known helium reserves were discovered by chance during oil and gas exploration, geologist Diveena Danabalan of Durham University in England and colleagues applied geologic know-how to their helium hunt. Helium accumulates underground during the radioactive decay of unstable elements such as uranium. That helium, though initially trapped, can be liberated when surrounding rock melts during volcanic activity. Using this information as well as seismic imaging of gas-trapping underground formations, the researchers discovered five spots in a volcanic region of Tanzania where water and helium-rich gas bubble to the surface from underground reservoirs.
The researchers predicted that they will be able to find more helium reservoirs and help meet society’s helium needs. Those needs go beyond just making balloons float and voices sound squeaky: Helium is essential for scientific research and a critical component of the cooling systems that allow medical MRI scanners to function.
All systems are go for the Juno spacecraft’s July 4 encounter with Jupiter.
“We couldn’t be more excited about being this close to Jupiter’s doorstep,” said Diane Brown, Juno program executive at NASA Headquarters in Washington, D.C., during a June 30 news briefing.
The scientific instruments have been shut off and the final command sequence for going into orbit around Jupiter has been uploaded to the spacecraft’s computers. On July 4, the probe will fire its main engine for 35 minutes, using it as a brake to slow down and be captured by Jupiter’s gravity. Once in orbit, Juno will spend 20 months figuring out what’s hiding beneath the thick clouds that encase the planet.
Juno has been busy during its final approach. On June 28, it got one more look at Jupiter and three of its moons. And last week Juno monitored changes in interplanetary plasma (see below) as it crossed a magnetic boundary that shields Jupiter from the stream of charged particles blowing from the sun.
Now all scientists can do is wait. “I have mixed emotions,” said mission lead Scott Bolton, a planetary scientist at the Southwest Research Institute in San Antonio. “I’m excited, but I also have tension and nervousness.” Juno has to perform a critical engine burn all on its own while passing through treacherous belts of radiation that encircle the planet. A series of radio tones from the spacecraft will let mission scientists know whether or not it worked.